11 Standard K-fold Cross-validation
Context and Purpose:
Upstream: Section @ref() -
Downstream:
Inputs:
Expected outputs:
In this section we will run K-fold cross-validation to evaluate the accuracy of predicting the performance of candidate parents (GEBV) who have not been phenotyped.
This is always recommended, and there are alternative kinds of predictions that could be set-up to measure this.
Important is the distinction from analyses we will do downstream to assess the accuracy of predicting the performance of crosses (i.e. mates).
We will use the runCrossVal() function.
We will demonstrate a few of the additional features that it provides in the process:
- Support for multiple traits
- Computing selection index accuracy
Finally, we’ll make a simple plot of the results.
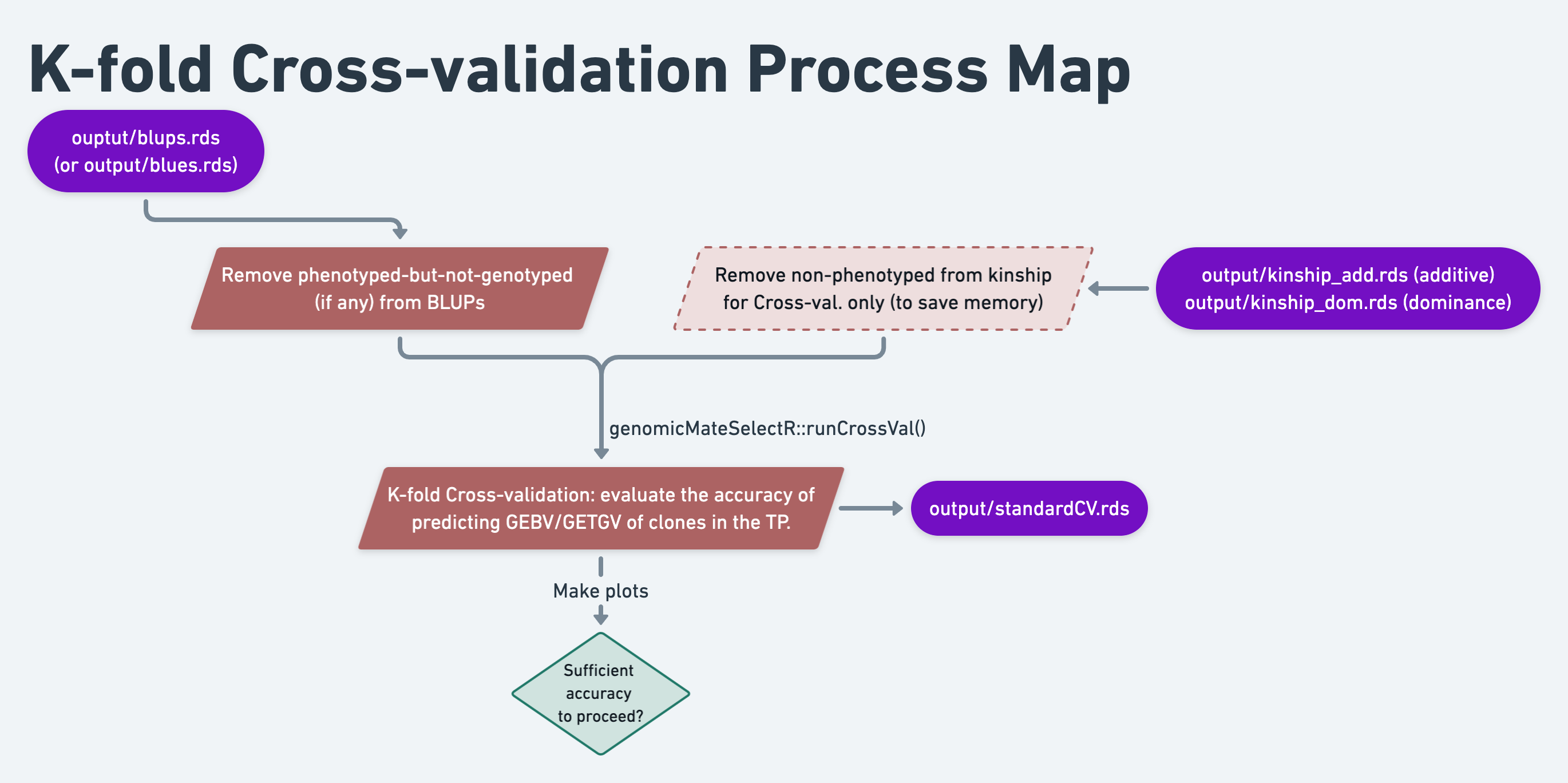
11.2 Set-up for the cross-validation
blups<-readRDS(here::here("output","blups.rds"))
A<-readRDS(file=here::here("output","kinship_add.rds"))
blups %<>%
# need to rename the "blups" list to comply with the runCrossVal function
rename(TrainingData=blups) %>%
dplyr::select(Trait,TrainingData) %>%
# need also to remove phenotyped-but-not-genotyped lines
# couldn't hurt to also subset the kinship to only phenotyped lines... would save RAM
mutate(TrainingData=map(TrainingData,
~filter(.,germplasmName %in% rownames(A)) %>%
# rename the germplasmName column to GID
rename(GID=germplasmName)))
blups
#> # A tibble: 4 × 2
#> Trait TrainingData
#> <chr> <list>
#> 1 DM <tibble [346 × 6]>
#> 2 MCMDS <tibble [292 × 6]>
#> 3 logFYLD <tibble [350 × 6]>
#> 4 logDYLD <tibble [348 × 6]>The steps above set-us up almost all the way.
11.3 Selection indices
Last thing: Let’s include selection index weights. You can find an excellent, detailed, open-source chapter from Walsh & Lynch on Selection Index Theory by clicking here.
\[SI = WT_1 \times Trait_1 + \dots + WT_t \times Trait_t\] Or in vector form:
\[SI = \boldsymbol{\hat{g}b}\]
\(SI\) is the selection index, dimension \([n \times 1]\). \(b\) is a \([t \times 1]\) vector of selection index “economic weights” designed to value each trait relative to its impact on the economic potential of changing the corresponding trait by one unit. Finally, \(\boldsymbol{\hat{g}\) is a matrix \([n \times t]\) with the (in this case) GEBV for each trait on the columns.
runCrossVal() will accept a named vector of selection index weights where names must match the “Trait” variable in blups using the SIwts= argument and setting selInd=TRUE.
Here are example weights, I’ll use. These are not to be taken as canonical. Weights should be determined for each target population of environments and product profile!
# I chose to remove MCMDS
## our preliminary analysis showed it to have ~0 heritability in this dataset
## initial test of cross-val. showed the models do not fit
SIwts<-c(DM=15,
#MCMDS=-10,
logFYLD=20,
logDYLD=20)
SIwts
#> DM logFYLD logDYLD
#> 15 20 20I’ll run a meager 2 repetitions of 5-fold cross-validation, which means 10 predictions per trait overall. I’ve got a 16-core laptop so I can use ncores=10 to do all 10 predictions per trait at the same time. runCrossVal() will process all four traits and compute the selection index accuracy at the end.
11.4 Execute cross-validation
starttime<-proc.time()[3]
standardCV<-runCrossVal(blups=blups %>% filter(Trait != "MCMDS"),
modelType="A",
selInd=TRUE,SIwts=SIwts,
grms=list(A=A),
nrepeats=2,nfolds=5,
gid="GID",seed=424242,
ncores=10)
#> Loading required package: rsample
#> Loading required package: furrr
#> Loading required package: future
#> iteration LogLik wall cpu(sec) restrained
#> 1 -150.932 9:29:44 0 0
#> 2 -150.587 9:29:44 0 0
#> 3 -150.456 9:29:44 0 0
#> 4 -150.431 9:29:44 0 0
#> 5 -150.429 9:29:45 1 0
#> 6 -150.429 9:29:45 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -109.584 9:29:45 0 0
#> 2 -109.57 9:29:45 0 0
#> 3 -109.562 9:29:45 0 0
#> 4 -109.56 9:29:45 0 0
#> 5 -109.559 9:29:45 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -115.829 9:29:45 0 0
#> 2 -115.829 9:29:46 1 0
#> 3 -115.828 9:29:46 1 0
#> 4 -115.828 9:29:46 1 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -153.247 9:29:45 0 0
#> 2 -153.244 9:29:45 0 0
#> 3 -153.243 9:29:45 0 0
#> 4 -153.243 9:29:45 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -108.226 9:29:45 0 0
#> 2 -108.147 9:29:45 0 0
#> 3 -108.101 9:29:45 0 0
#> 4 -108.087 9:29:46 1 0
#> 5 -108.085 9:29:46 1 0
#> 6 -108.085 9:29:46 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -117.592 9:29:46 0 0
#> 2 -117.537 9:29:46 0 0
#> 3 -117.513 9:29:46 0 0
#> 4 -117.509 9:29:46 0 0
#> 5 -117.508 9:29:46 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -150.198 9:29:45 0 0
#> 2 -149.363 9:29:45 0 0
#> 3 -148.987 9:29:46 1 0
#> 4 -148.881 9:29:46 1 0
#> 5 -148.865 9:29:46 1 0
#> 6 -148.863 9:29:46 1 0
#> 7 -148.862 9:29:46 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -106.107 9:29:46 0 0
#> 2 -105.581 9:29:46 0 0
#> 3 -105.152 9:29:46 0 0
#> 4 -104.92 9:29:46 0 0
#> 5 -104.852 9:29:46 0 0
#> 6 -104.832 9:29:47 1 0
#> 7 -104.827 9:29:47 1 0
#> 8 -104.825 9:29:47 1 0
#> 9 -104.825 9:29:47 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -118.481 9:29:47 0 0
#> 2 -118.255 9:29:47 0 0
#> 3 -118.106 9:29:47 0 0
#> 4 -118.047 9:29:47 0 0
#> 5 -118.035 9:29:47 0 0
#> 6 -118.032 9:29:47 0 0
#> 7 -118.032 9:29:47 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -144.958 9:29:46 0 0
#> 2 -144.946 9:29:46 0 0
#> 3 -144.94 9:29:46 0 0
#> 4 -144.939 9:29:46 0 0
#> 5 -144.939 9:29:46 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -107.241 9:29:47 1 0
#> 2 -107.24 9:29:47 1 0
#> 3 -107.24 9:29:47 1 0
#> 4 -107.24 9:29:47 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -114.776 9:29:47 0 0
#> 2 -114.775 9:29:47 0 0
#> 3 -114.775 9:29:47 0 0
#> 4 -114.775 9:29:47 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -150.502 9:29:47 1 0
#> 2 -150.404 9:29:47 1 0
#> 3 -150.354 9:29:47 1 0
#> 4 -150.339 9:29:47 1 0
#> 5 -150.336 9:29:47 1 0
#> 6 -150.336 9:29:47 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -112.48 9:29:47 0 0
#> 2 -112.42 9:29:47 0 0
#> 3 -112.38 9:29:47 0 0
#> 4 -112.364 9:29:47 0 0
#> 5 -112.36 9:29:48 1 0
#> 6 -112.358 9:29:48 1 0
#> 7 -112.358 9:29:48 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -118.347 9:29:48 0 0
#> 2 -118.041 9:29:48 0 0
#> 3 -117.869 9:29:48 0 0
#> 4 -117.803 9:29:48 0 0
#> 5 -117.787 9:29:48 0 0
#> 6 -117.784 9:29:48 0 0
#> 7 -117.783 9:29:48 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -150.226 9:29:47 0 0
#> 2 -149.466 9:29:47 0 0
#> 3 -149.138 9:29:47 0 0
#> 4 -149.063 9:29:47 0 0
#> 5 -149.056 9:29:47 0 0
#> 6 -149.055 9:29:47 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -111.205 9:29:48 0 0
#> 2 -111.2 9:29:48 0 0
#> 3 -111.196 9:29:48 0 0
#> 4 -111.193 9:29:48 0 0
#> 5 -111.193 9:29:48 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -115.15 9:29:48 0 0
#> 2 -115.132 9:29:48 0 0
#> 3 -115.119 9:29:48 0 0
#> 4 -115.114 9:29:49 1 0
#> 5 -115.113 9:29:49 1 0
#> 6 -115.112 9:29:49 1 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -150.983 9:29:48 0 0
#> 2 -150.511 9:29:48 0 0
#> 3 -150.265 9:29:48 0 0
#> 4 -150.179 9:29:48 0 0
#> 5 -150.162 9:29:48 0 0
#> 6 -150.158 9:29:48 0 0
#> 7 -150.157 9:29:48 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -109.264 9:29:48 0 0
#> 2 -109.264 9:29:48 0 0
#> 3 -109.264 9:29:49 1 0
#> 4 -109.263 9:29:49 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -116.271 9:29:49 0 0
#> 2 -116.238 9:29:49 0 0
#> 3 -116.225 9:29:49 0 0
#> 4 -116.223 9:29:49 0 0
#> 5 -116.223 9:29:49 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -146.729 9:29:48 0 0
#> 2 -146.707 9:29:48 0 0
#> 3 -146.695 9:29:48 0 0
#> 4 -146.691 9:29:48 0 0
#> 5 -146.691 9:29:49 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -105.14 9:29:49 0 0
#> 2 -105.116 9:29:49 0 0
#> 3 -105.101 9:29:49 0 0
#> 4 -105.095 9:29:49 0 0
#> 5 -105.095 9:29:49 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -116.469 9:29:49 0 0
#> 2 -116.439 9:29:49 0 0
#> 3 -116.428 9:29:49 0 0
#> 4 -116.426 9:29:50 1 0
#> 5 -116.426 9:29:50 1 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -146.167 9:29:49 0 0
#> 2 -145.784 9:29:49 0 0
#> 3 -145.645 9:29:49 0 0
#> 4 -145.618 9:29:49 0 0
#> 5 -145.616 9:29:49 0 0
#> 6 -145.616 9:29:49 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -108.335 9:29:49 0 0
#> 2 -108.255 9:29:49 0 0
#> 3 -108.205 9:29:50 1 0
#> 4 -108.187 9:29:50 1 0
#> 5 -108.184 9:29:50 1 0
#> 6 -108.184 9:29:50 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -115.606 9:29:50 0 0
#> 2 -115.563 9:29:50 0 0
#> 3 -115.541 9:29:50 0 0
#> 4 -115.535 9:29:50 0 0
#> 5 -115.534 9:29:50 0 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -152 9:29:49 0 0
#> 2 -151.698 9:29:49 0 0
#> 3 -151.579 9:29:49 0 0
#> 4 -151.555 9:29:50 1 0
#> 5 -151.554 9:29:50 1 0
#> 6 -151.553 9:29:50 1 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -107.98 9:29:50 0 0
#> 2 -107.972 9:29:50 0 0
#> 3 -107.968 9:29:50 0 0
#> 4 -107.967 9:29:50 0 0
#> [1] "GBLUP model complete - one trait"
#> iteration LogLik wall cpu(sec) restrained
#> 1 -119.501 9:29:50 0 0
#> 2 -119.452 9:29:50 0 0
#> 3 -119.431 9:29:50 0 0
#> 4 -119.426 9:29:51 1 0
#> 5 -119.425 9:29:51 1 0
#> [1] "GBLUP model complete - one trait"
#> [1] "Genomic predictions done for all traits in one repeat-fold"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
#> Joining, by = "GID"
timeelapsed<-proc.time()[3]-starttime;
timeelapsed/60
#> elapsed
#> 0.1538Save the results
11.5 Plot results
standardCV %>%
unnest(accuracyEstOut) %>%
dplyr::select(repeats,id,predOf,Trait,Accuracy) %>%
ggplot(.,aes(x=Trait,y=Accuracy,fill=Trait)) +
geom_boxplot() + theme_bw()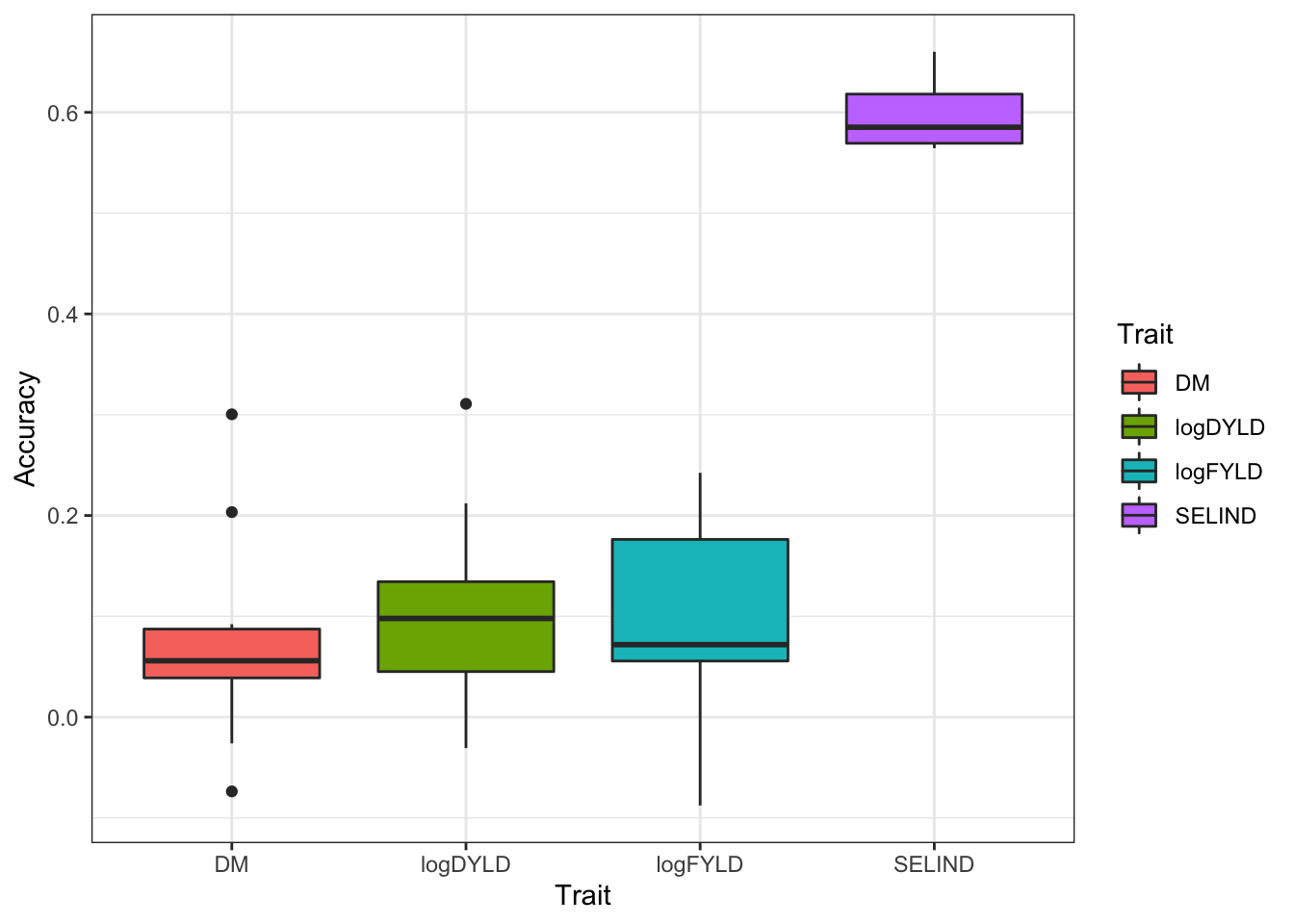
This result is not what I would expect. SELIND should be similar to individual trait accuracies.
Best guess: SELIND requires the BLUPs for each trait to be observed, so only the clones with complete data will be included.
Your results will be different if you choose a different dataset, hopefully better.