Pre-process data files for downstream cross-validation
Marnin Wolfe
2021-May-13
Last updated: 2021-07-29
Checks: 7 0
Knit directory: implementGMSinCassava/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210504) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version bc85a7d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/accuracies.png
Ignored: analysis/fig2.png
Ignored: analysis/fig3.png
Ignored: analysis/fig4.png
Ignored: code/.DS_Store
Ignored: data/.DS_Store
Untracked files:
Untracked: accuracies.png
Untracked: analysis/docs/
Untracked: analysis/speedUpPredCrossVar.Rmd
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.bed
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.bim
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.fam
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.hap.gz
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.log
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.nosex
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.sample
Untracked: data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719.vcf.gz
Untracked: data/DatabaseDownload_2021May04/
Untracked: data/blups_forCrossVal.rds
Untracked: data/config.txt
Untracked: data/config_mw.txt
Untracked: data/dosages_IITA_2021May13.rds
Untracked: data/dosages_IITA_filtered_2021May13.rds
Untracked: data/genmap_2021May13.rds
Untracked: data/haps_IITA_2021May13.rds
Untracked: data/haps_IITA_filtered_2021May13.rds
Untracked: data/recombFreqMat_1minus2c_2021May13.rds
Untracked: fig2.png
Untracked: fig3.png
Untracked: figure/
Untracked: output/Cassava_HMII_V3_Marning_imputation_6-18-21.samples
Untracked: output/IITA_CleanedTrialData_2021May10.rds
Untracked: output/IITA_ExptDesignsDetected_2021May10.rds
Untracked: output/IITA_blupsForModelTraining_twostage_asreml_2021May10.rds
Untracked: output/IITA_trials_NOT_identifiable.csv
Untracked: output/alphaAssignOutput_iita_pedigree.txt.dams
Untracked: output/alphaAssignOutput_iita_pedigree.txt.dams.full
Untracked: output/alphaAssignOutput_iita_pedigree.txt.pedigree
Untracked: output/alphaAssignOutput_iita_pedigree.txt.pedigree.top
Untracked: output/alphaAssignOutput_iita_pedigree.txt.sires
Untracked: output/alphaAssignOutput_iita_pedigree.txt.sires.full
Untracked: output/crossValPredsA.rds
Untracked: output/crossValPredsAD.rds
Untracked: output/cvAD_1rep_markerEffects.rds
Untracked: output/cvAD_1rep_meanPredAccuracy.rds
Untracked: output/cvAD_1rep_parentfolds.rds
Untracked: output/cvAD_1rep_predAccuracy.rds
Untracked: output/cvAD_1rep_predMeans.rds
Untracked: output/cvAD_1rep_predVars.rds
Untracked: output/cvAD_1rep_varPredAccuracy.rds
Untracked: output/cvAD_5rep5fold_markerEffects.rds
Untracked: output/cvAD_5rep5fold_meanPredAccuracy.rds
Untracked: output/cvAD_5rep5fold_parentfolds.rds
Untracked: output/cvAD_5rep5fold_predMeans.rds
Untracked: output/cvAD_5rep5fold_predVars.rds
Untracked: output/cvAD_5rep5fold_varPredAccuracy.rds
Untracked: output/cvDirDom_5rep5fold_markerEffects.rds
Untracked: output/cvDirDom_5rep5fold_meanPredAccuracy.rds
Untracked: output/cvDirDom_5rep5fold_parentfolds.rds
Untracked: output/cvDirDom_5rep5fold_predMeans.rds
Untracked: output/cvDirDom_5rep5fold_predVars.rds
Untracked: output/cvDirDom_5rep5fold_varPredAccuracy.rds
Untracked: output/cvMeanPredAccuracyA.rds
Untracked: output/cvMeanPredAccuracyAD.rds
Untracked: output/cvPredMeansA.rds
Untracked: output/cvPredMeansAD.rds
Untracked: output/cvVarPredAccuracyA.rds
Untracked: output/cvVarPredAccuracyAD.rds
Untracked: output/genomicMatePredictions_top121parents_ModelAD.csv
Untracked: output/genomicMatePredictions_top121parents_ModelAD.rds
Untracked: output/genomicMatePredictions_top121parents_ModelDirDom.csv
Untracked: output/genomicMatePredictions_top121parents_ModelDirDom.rds
Untracked: output/genomicPredictions_ModelAD.csv
Untracked: output/genomicPredictions_ModelAD.rds
Untracked: output/genomicPredictions_ModelDirDom.csv
Untracked: output/genomicPredictions_ModelDirDom.rds
Untracked: output/kinship_A_IITA_2021May13.rds
Untracked: output/kinship_D_IITA_2021May13.rds
Untracked: output/kinship_domGenotypic_IITA_2021July5.rds
Untracked: output/markEffsTest.rds
Untracked: output/markerEffects.rds
Untracked: output/markerEffectsA.rds
Untracked: output/markerEffectsAD.rds
Untracked: output/maxNOHAV_byStudy.csv
Untracked: output/obsCrossMeansAndVars.rds
Untracked: output/parentfolds.rds
Untracked: output/ped2check_genome.rds
Untracked: output/ped2genos.txt
Untracked: output/pednames2keep.txt
Untracked: output/pednames_Prune100_25_pt25.log
Untracked: output/pednames_Prune100_25_pt25.nosex
Untracked: output/pednames_Prune100_25_pt25.prune.in
Untracked: output/pednames_Prune100_25_pt25.prune.out
Untracked: output/potential_dams.txt
Untracked: output/potential_sires.txt
Untracked: output/predVarTest.rds
Untracked: output/samples2keep_IITA_2021May13.txt
Untracked: output/samples2keep_IITA_MAFpt01_prune50_25_pt98.log
Untracked: output/samples2keep_IITA_MAFpt01_prune50_25_pt98.nosex
Untracked: output/samples2keep_IITA_MAFpt01_prune50_25_pt98.prune.in
Untracked: output/samples2keep_IITA_MAFpt01_prune50_25_pt98.prune.out
Untracked: output/samples2keep_notInPHGdb.txt
Untracked: output/test_cvAD_markerEffects.rds
Untracked: output/test_cvAD_meanPredAccuracy.rds
Untracked: output/test_cvAD_parentfolds.rds
Untracked: output/test_cvAD_predAccuracy.rds
Untracked: output/test_cvAD_predMeans.rds
Untracked: output/test_cvAD_predVars.rds
Untracked: output/test_cvAD_varPredAccuracy.rds
Untracked: output/top50crosses.csv
Untracked: output/verified_ped.txt
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/04-PreprocessDataFiles.Rmd) and HTML (docs/04-PreprocessDataFiles.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 934141c | wolfemd | 2021-07-14 | Build site. |
| Rmd | 04a2ca8 | wolfemd | 2021-07-05 | Add genotypic dominance matrix. |
| html | e66bdad | wolfemd | 2021-06-10 | Build site. |
| Rmd | a8452ba | wolfemd | 2021-06-10 | Initial build of the entire page upon completion of all |
Previous step
- Validate the pedigree obtained from cassavabase: Before setting up a cross-validation scheme for predictions that depend on a correct pedigree, add a basic verification step to the pipeline. Not trying to fill unknown or otherwise correct the pedigree. Assess evidence that relationship is correct, remove if incorrect.
Haplotype matrix from phased VCF
Extract haps from VCF with bcftools
library(tidyverse); library(magrittr)
pathIn<-"/home/jj332_cas/marnin/implementGMSinCassava/data/"
pathOut<-pathIn
vcfName<-"AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719"
system(paste0("bcftools convert --hapsample ",
pathOut,vcfName," ",
pathIn,vcfName,".vcf.gz "))Read haps to R
library(data.table)
haps<-fread(paste0(pathIn,vcfName,".hap.gz"),
stringsAsFactors = F,header = F) %>%
as.data.frame
sampleids<-fread(paste0(pathIn,vcfName,".sample"),
stringsAsFactors = F,header = F,skip = 2) %>%
as.data.frameExtract needed GIDs from BLUPs and pedigree: Subset to: (1) genotyped-plus-phenotyped and/or (2) in verified pedigree.
blups<-readRDS(file=here::here("output",
"IITA_blupsForModelTraining_twostage_asreml_2021May10.rds"))
blups %>%
select(Trait,blups) %>%
unnest(blups) %>%
distinct(GID) %$% GID -> gidWithBLUPs
genotypedWithBLUPs<-gidWithBLUPs[gidWithBLUPs %in% sampleids$V1]
length(genotypedWithBLUPs) # 7960
ped<-read.table(here::here("output","verified_ped.txt"),
header = T, stringsAsFactors = F)
pednames<-union(ped$FullSampleName,
union(ped$SireID,ped$DamID))
length(pednames) # 4384
samples2keep<-union(genotypedWithBLUPs,pednames)
length(samples2keep) # 8013
# write a sample list to disk for downstream purposes
# format suitable for subsetting with --keep in plink
write.table(tibble(FID=0,IID=samples2keep),
file=here::here("output","samples2keep_IITA_2021May13.txt"),
row.names = F, col.names = F, quote = F)Add sample ID’s
hapids<-sampleids %>%
select(V1,V2) %>%
mutate(SampleIndex=1:nrow(.)) %>%
rename(HapA=V1,HapB=V2) %>%
pivot_longer(cols=c(HapA,HapB),
names_to = "Haplo",values_to = "SampleID") %>%
mutate(HapID=paste0(SampleID,"_",Haplo)) %>%
arrange(SampleIndex)
colnames(haps)<-c("Chr","HAP_ID","Pos","REF","ALT",hapids$HapID)Subset haps
hapids2keep<-hapids %>% filter(SampleID %in% samples2keep)
hapids2keep$HapID
dim(haps) # [1] 68814 43717
haps<-haps[,c("Chr","HAP_ID","Pos","REF","ALT",hapids2keep$HapID)]
dim(haps) # [1] 68814 16031Format, transpose, convert to matrix and save!
haps %<>%
mutate(HAP_ID=gsub(":","_",HAP_ID)) %>%
column_to_rownames(var = "HAP_ID") %>%
select(-Chr,-Pos,-REF,-ALT)
haps %<>% t(.) %>% as.matrix(.)
saveRDS(haps,file=here::here("data","haps_IITA_2021May13.rds")Make dosages from haps
To ensure consistency in allele counting, create dosage from haps manually.
dosages<-haps %>%
as.data.frame(.) %>%
rownames_to_column(var = "GID") %>%
separate(GID,c("SampleID","Haplo"),"_Hap",remove = T) %>%
select(-Haplo) %>%
group_by(SampleID) %>%
summarise(across(everything(),~sum(.))) %>%
ungroup() %>%
column_to_rownames(var = "SampleID") %>%
as.matrix
saveRDS(dosages,file=here::here("data","dosages_IITA_2021May13.rds"))
# > dim(dosages)
# [1] 8013 68814Variant filters
Apply a MAF filter and lightly LD prune: The number of markers in the “raw” dataset (~68K) is ~3X the number used in the mate selection paper and I think more than is necessary. There is a burden incurred because we have to compute and store in memory (and on disk) \(N_{snp} \times N_{snp}\) recombination frequency matrices.
# library(tidyverse); library(magrittr)
# pathIn<-"/home/jj332_cas/marnin/implementGMSinCassava/data/"
# pathOut<-pathIn
# vcfName<-"AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719"
#
# write.table(tibble(FID=0,IID=samples2keep),
# file=here::here("output","samples2keep_IITA_2021May13.txt"),
# row.names = F, col.names = F, quote = F)
#
# ped2check<-read.table(file=here::here("output","ped2genos.txt"),
# header = F, stringsAsFactors = F)
#
# # pednames<-union(ped2check$V1,union(ped2check$V2,ped2check$V3)) %>%
# # tibble(FID=0,IID=.)
# # write.table(pednames,file=here::here("output","pednames2keep.txt"),
# # row.names = F, col.names = F, quote = F)cd /home/jj332_cas/marnin/implementGMSinCassava/
export PATH=/programs/plink-1.9-x86_64-beta3.30:$PATH;
plink --bfile data/AllChrom_RefPanelAndGSprogeny_ReadyForGP_72719 \
--keep output/samples2keep_IITA_2021May13.txt \
--maf 0.01 \
--indep-pairwise 50 25 0.98 \
--out output/samples2keep_IITA_MAFpt01_prune50_25_pt98;Used plink to output a list of pruned SNPs.
Next, subset the columns of haps and dosages in R.
library(tidyverse); library(magrittr);
haps<-readRDS(file=here::here("data","haps_IITA_2021May13.rds"))
dosages<-readRDS(file=here::here("data","dosages_IITA_2021May13.rds"))
snps2keep<-read.table(here::here("output",
"samples2keep_IITA_MAFpt01_prune50_25_pt98.prune.in"),
header = F, stringsAsFactors = F)
snps2keep<-tibble(HapSNP_ID=colnames(haps)) %>%
separate(HapSNP_ID,c("Chr","Pos","Ref","Alt"),remove = F) %>%
mutate(SNP_ID=paste0("S",Chr,"_",Pos)) %>%
filter(SNP_ID %in% snps2keep$V1)
haps<-haps[,snps2keep$HapSNP_ID]
dosages<-dosages[,snps2keep$HapSNP_ID]
# dim(haps); dim(dosages); haps[1:5,1:10]
saveRDS(haps,file=here::here("data","haps_IITA_filtered_2021May13.rds"))
saveRDS(dosages,file=here::here("data","dosages_IITA_filtered_2021May13.rds"))Make Add and Dom GRMs from dosages
# activate multithread OpenBLAS for fast matrix algebra
export OMP_NUM_THREADS=56dosages<-readRDS(file=here::here("data","dosages_IITA_filtered_2021May13.rds"))
A<-predCrossVar::kinship(dosages,type="add")
D<-predCrossVar::kinship(dosages,type="dom")
saveRDS(A,file=here::here("output","kinship_A_IITA_2021May13.rds"))
saveRDS(D,file=here::here("output","kinship_D_IITA_2021May13.rds"))cd /home/mw489/implementGMSinCassava/;
screen;
singularity shell rocker.sif; Rdosages<-readRDS(file=here::here("data","dosages_IITA_filtered_2021May13.rds"))
source(here::here("code","gsFunctions.R"))
RhpcBLASctl::blas_set_num_threads(56)
D<-kinship(dosages,type="domGenotypic")
saveRDS(D,file=here::here("output","kinship_domGenotypic_IITA_2021July5.rds"))Genetic Map
cp -r /home/jj332_cas/CassavaGenotypeData/CassavaGeneticMap /home/jj332_cas/marnin/implementGMSinCassava/data/# activate multithread OpenBLAS for fast matrix algebra
export OMP_NUM_THREADS=56Creating the map used for Beagle-imputation in 2019: In 2019, I obtained a ICGMC-derived genetic map, I think from Guillaume Bauchet and used it to create a map I’ve been using for imputation, which has 25K markers (Beagle interpolates the map to the markers genotyped in the panel).
However, the recombination frequency matrix and thus cross-variance predictions needs to have all positions for which we have marker effects. It means I have to interpolate a map from the original file cassava_cM_pred.v6.allchr.txt. See below:
library(tidyverse); library(magrittr)
dosages<-readRDS(file=here::here("data","dosages_IITA_filtered_2021May13.rds"))
# genmap<-tibble(Chr=1:18) %>%
# mutate(geneticMap=map(Chr,~read.table(here::here("data/CassavaGeneticMap",
# paste0("chr",.,"_cassava_cM_pred.v6_91019.map")),
# header = F, stringsAsFactors = F)))
genmap<-read.table(here::here("data/CassavaGeneticMap",
"cassava_cM_pred.v6.allchr.txt"),
header = F, stringsAsFactors = F,sep=';') %>%
rename(SNP_ID=V1,Pos=V2,cM=V3) %>%
as_tibble
snps_genmap<-tibble(DoseSNP_ID=colnames(dosages)) %>%
separate(DoseSNP_ID,c("Chr","Pos","Ref","Alt"),remove = F) %>%
mutate(SNP_ID=paste0("S",Chr,"_",Pos)) %>%
left_join(genmap %>% mutate(across(everything(),as.character)))
# snps_genmap %>%
# ggplot(.,aes(x=as.integer(Pos),y=as.numeric(cM))) +
# geom_point() +
# theme_bw() +
# facet_wrap(~Chr)interpolate_genmap<-function(data){
# for each chromosome map
# find and _decrements_ in the genetic map distance
# fix them to the cumulative max to force map to be only increasing
# fit a spline for each chromosome
# Use it to predict values for positions not previously on the map
# fix them AGAIN (in case) to the cumulative max, forcing map to only increase
data_forspline<-data %>%
filter(!is.na(cM)) %>%
mutate(cumMax=cummax(cM),
cumIncrement=cM-cumMax) %>%
filter(cumIncrement>=0) %>%
select(-cumMax,-cumIncrement)
spline<-data_forspline %$% smooth.spline(x=Pos,y=cM,spar = 0.75)
splinemap<-predict(spline,x = data$Pos) %>%
as_tibble(.) %>%
rename(Pos=x,cM=y) %>%
mutate(cumMax=cummax(cM),
cumIncrement=cM-cumMax) %>%
mutate(cM=cumMax) %>%
select(-cumMax,-cumIncrement)
return(splinemap)
}splined_snps_genmap<-snps_genmap %>%
select(-cM) %>%
mutate(Pos=as.numeric(Pos)) %>%
left_join(snps_genmap %>%
mutate(across(c(Pos,cM),as.numeric)) %>%
arrange(Chr,Pos) %>%
nest(-Chr) %>%
mutate(data=map(data,interpolate_genmap)) %>%
unnest(data)) %>%
distinctall(splined_snps_genmap$DoseSNP_ID == colnames(dosages))[1] TRUE# [1] TRUE
saveRDS(splined_snps_genmap,file=here::here("data","genmap_2021May13.rds"))splined_snps_genmap %>%
mutate(Map="Spline") %>%
bind_rows(snps_genmap %>%
mutate(across(c(Pos,cM),as.numeric)) %>%
arrange(Chr,Pos) %>% mutate(Map="Data")) %>%
ggplot(.,aes(x=Pos,y=cM,color=Map),alpha=0.5,size=0.75) +
geom_point() +
theme_bw() + facet_wrap(~as.integer(Chr), scales='free_x')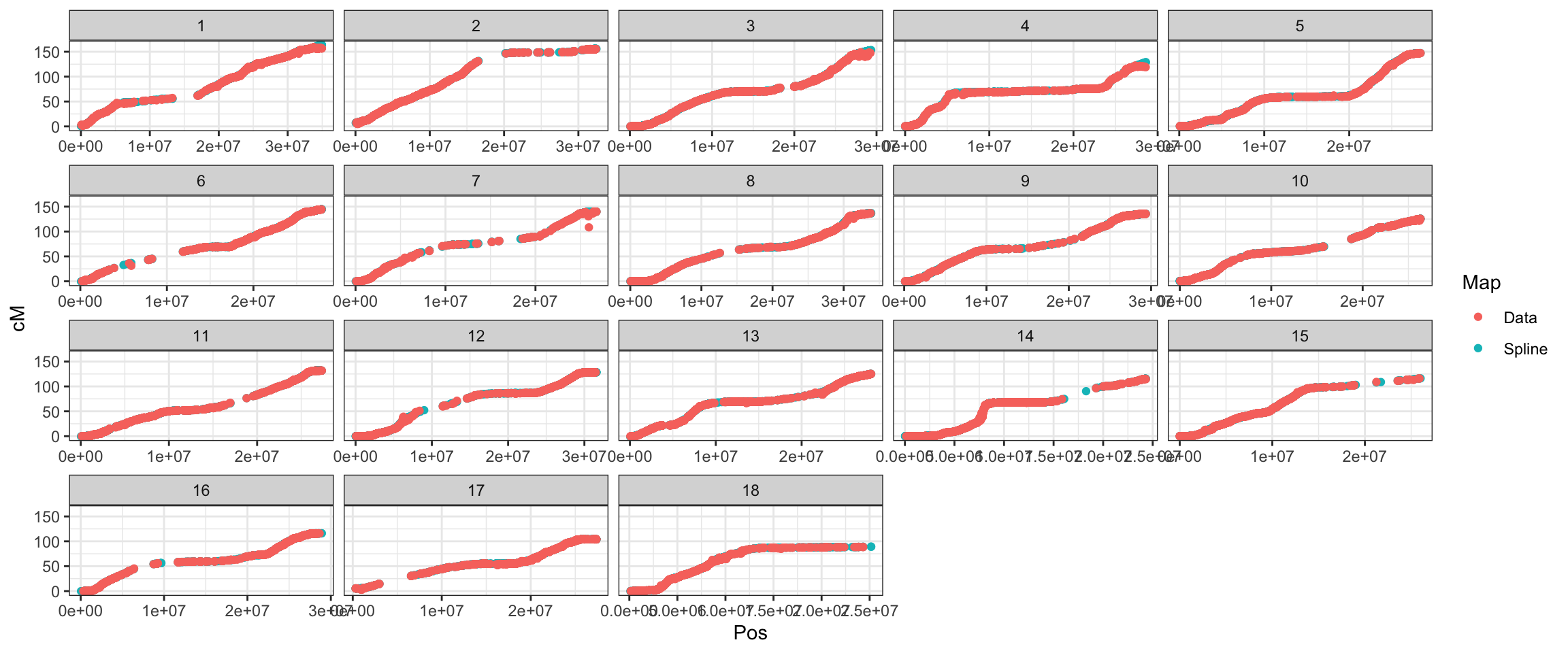
| Version | Author | Date |
|---|---|---|
| 934141c | wolfemd | 2021-07-14 |
Recomb. freq. matrix
Construct a matrix of recombination frequencies at loci for all study loci. Pre-compute 1-2c to save time predicting cross variance.
library(predCrossVar)
genmap<-readRDS(file=here::here("data","genmap_2021May13.rds"))
m<-genmap$cM;
names(m)<-genmap$DoseSNP_ID
recombFreqMat<-1-(2*genmap2recombfreq(m,nChr = 18))
saveRDS(recombFreqMat,file=here::here("data","recombFreqMat_1minus2c_2021May13.rds"))Pick traits to cross-validate
# This list from Dec. 2020 GeneticGain rate estimation
# These were what Ismail/IITA/BMGF wanted to see
# Will cross-validate these traits
traits<-c("logDYLD","logFYLD","logRTNO","logTOPYLD","MCMDS","DM","BCHROMO",
"PLTHT","BRLVLS","BRNHT1","HI")
# Full trait list = 14:
## traits<-c("MCMDS","DM","PLTHT","BRNHT1","BRLVLS","HI",
## "logDYLD", # <-- logDYLD now included.
## "logFYLD","logTOPYLD","logRTNO","TCHART","LCHROMO","ACHROMO","BCHROMO")Pedigree
- Validate the pedigree obtained from cassavabase: Before setting up a cross-validation scheme for predictions that depend on a correct pedigree, add a basic verification step to the pipeline. Not trying to fill unknown or otherwise correct the pedigree. Assess evidence that relationship is correct, remove if incorrect.
ped<-read.table(here::here("output","verified_ped.txt"),
header = T, stringsAsFactors = F)BLUPs
Select traits and data to be analyzed.
library(tidyverse); library(magrittr);
blups<-readRDS(file=here::here("output",
"IITA_blupsForModelTraining_twostage_asreml_2021May10.rds"))
dosages<-readRDS(file=here::here("data","dosages_IITA_filtered_2021May13.rds"))
blups %>%
select(Trait,blups) %>%
unnest(blups) %>%
distinct(GID) %$% GID -> gidWithBLUPs
genotypedWithBLUPs<-gidWithBLUPs[gidWithBLUPs %in% rownames(dosages)]
length(genotypedWithBLUPs) # 7960
blups %<>%
filter(Trait %in% traits) %>%
select(Trait,blups,varcomp) %>%
mutate(blups=map(blups,~filter(.,GID %in% genotypedWithBLUPs)))
saveRDS(blups,file=here::here("data","blups_forCrossVal.rds"))Index weights [get from Ismail]
# library(tidyverse); library(magrittr); library(predCrossVar); library(BGLR);
# blups<-readRDS(here::here("data","blups_forawcdata.rds")) %>%
# select(Trait,blups) %>% # BLUPs long-->wide for multivar analysis
# unnest(blups) %>%
# select(Trait,germplasmName,drgBLUP) %>%
# spread(Trait,drgBLUP)
#
# indices<-blups %>%
# summarize_if(is.numeric,sd, na.rm=T) %>%
# pivot_longer(cols = everything(), names_to = "Trait", values_to = "blupSD") %>%
# left_join(tibble(Trait=c("DM","logFYLD","MCMDS","TCHART"),
# stdSI_unscaled=c(5, 10, -10, -5),
# biofortSI_unscaled=c(10, 5, -5,10))) %>%
# mutate(stdSI=stdSI_unscaled/blupSD,
# biofortSI=biofortSI_unscaled/blupSD)
# indices %>% mutate_if(is.numeric,~round(.,2))
# saveRDS(indices,file=here::here("data","selection_index_weights_4traits.rds"))Next step
- Parent-wise cross-validation: Compute parent-wise cross-validation folds using the validated pedigree. Fit models to get marker effects and make subsequent predictions of cross means and (co)variances.
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] magrittr_2.0.1 forcats_0.5.1 stringr_1.4.0 dplyr_1.0.7
[5] purrr_0.3.4 readr_2.0.0 tidyr_1.1.3 tibble_3.1.3
[9] ggplot2_3.3.5 tidyverse_1.3.1 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 lubridate_1.7.10 here_1.0.1 assertthat_0.2.1
[5] rprojroot_2.0.2 digest_0.6.27 utf8_1.2.2 R6_2.5.0
[9] cellranger_1.1.0 backports_1.2.1 reprex_2.0.0 evaluate_0.14
[13] highr_0.9 httr_1.4.2 pillar_1.6.2 rlang_0.4.11
[17] readxl_1.3.1 rstudioapi_0.13 whisker_0.4 jquerylib_0.1.4
[21] rmarkdown_2.9 labeling_0.4.2 munsell_0.5.0 broom_0.7.9
[25] compiler_4.1.0 httpuv_1.6.1 modelr_0.1.8 xfun_0.24
[29] pkgconfig_2.0.3 htmltools_0.5.1.1 tidyselect_1.1.1 fansi_0.5.0
[33] crayon_1.4.1 tzdb_0.1.2 dbplyr_2.1.1 withr_2.4.2
[37] later_1.2.0 grid_4.1.0 jsonlite_1.7.2 gtable_0.3.0
[41] lifecycle_1.0.0 DBI_1.1.1 git2r_0.28.0 scales_1.1.1
[45] cli_3.0.1 stringi_1.7.3 farver_2.1.0 fs_1.5.0
[49] promises_1.2.0.1 xml2_1.3.2 bslib_0.2.5.1 ellipsis_0.3.2
[53] generics_0.1.0 vctrs_0.3.8 tools_4.1.0 glue_1.4.2
[57] hms_1.1.0 yaml_2.2.1 colorspace_2.0-2 rvest_1.0.1
[61] knitr_1.33 haven_2.4.1 sass_0.4.0