Using AlphaSimHlpR
Jean-Luc Jannink
2020-01-05
Using_AlphaSimHlpR.RmdAlphaSimR is a package with many useful functions to simulate plant and animal breeding schemes. But it is not very easy to implement. AlfSimHlpR defines functions that provide structure for a breeding scheme
Using AlfSimHlpR
# Make sure you have the right packages installed
neededPackages <- c("AlphaSimR", "dplyr", "tidyr", "plotrix", "lme4", "sommer", "optiSel")
for (p in neededPackages) if (!require(p, character.only=T)) install.packages(p)
#> Loading required package: AlphaSimR
#> Loading required package: R6
#> Loading required package: dplyr
#>
#> Attaching package: 'dplyr'
#> The following object is masked from 'package:AlphaSimR':
#>
#> mutate
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
#> Loading required package: tidyr
#> Loading required package: plotrix
#> Loading required package: lme4
#> Loading required package: Matrix
#>
#> Attaching package: 'Matrix'
#> The following objects are masked from 'package:tidyr':
#>
#> expand, pack, unpack
#> Loading required package: sommer
#> Loading required package: MASS
#>
#> Attaching package: 'MASS'
#> The following object is masked from 'package:dplyr':
#>
#> select
#> Loading required package: lattice
#> Loading required package: crayon
#> Loading required package: optiSel
suppressMessages(library(AlphaSimHlpR))Define simulation settings
Define the genetic architecture of the population and other breeding scheme parameters in a list bsp.
bsp <- specifyPopulation(ctrlFileName="../inst/PopulationCtrlFile_Small.txt")
bsp <- specifyPipeline(bsp, ctrlFileName="../inst/ControlFile_Small.txt")
bsp <- specifyCosts(bsp, ctrlFileName="../inst/CostsCtrlFile_Small.txt")
print(bsp)
#> $nChr
#> [1] 3
#>
#> $effPopSize
#> [1] 100
#>
#> $quickHaplo
#> [1] TRUE
#>
#> $segSites
#> [1] 400
#>
#> $nQTL
#> [1] 40
#>
#> $nSNP
#> [1] 100
#>
#> $genVar
#> [1] 40
#>
#> $gxeVar
#> numeric(0)
#>
#> $gxyVar
#> [1] 15
#>
#> $gxlVar
#> [1] 10
#>
#> $gxyxlVar
#> [1] 5
#>
#> $meanDD
#> [1] 0.8
#>
#> $varDD
#> [1] 0.01
#>
#> $relAA
#> [1] 0.5
#>
#> $nStages
#> [1] 3
#>
#> $stageNames
#> [1] "SDN" "CET" "PYT"
#>
#> $stageToGenotype
#> [1] "CET"
#>
#> $trainingPopCycles
#> F1 SDN CET PYT
#> 0 3 3 2
#>
#> $nParents
#> [1] 15
#>
#> $nCrosses
#> [1] 30
#>
#> $nProgeny
#> [1] 10
#>
#> $usePolycrossNursery
#> [1] FALSE
#>
#> $nSeeds
#> [1] 300
#>
#> $useOptContrib
#> [1] FALSE
#>
#> $nCandOptCont
#> [1] 200
#>
#> $targetEffPopSize
#> [1] 20
#>
#> $nEntries
#> SDN CET PYT
#> 200 75 20
#>
#> $nReps
#> SDN CET PYT
#> 1 1 2
#>
#> $nLocs
#> SDN CET PYT
#> 1 2 2
#>
#> $nClonesToNCRP
#> [1] 3
#>
#> $nChks
#> SDN CET PYT
#> 5 4 2
#>
#> $entryToChkRatio
#> SDN CET PYT
#> 50 25 20
#>
#> $errVars
#> SDN CET PYT
#> 200 100 70
#>
#> $phenoF1toStage1
#> [1] TRUE
#>
#> $errVarPreStage1
#> [1] 500
#>
#> $useCurrentPhenoTrain
#> [1] FALSE
#>
#> $nCyclesToKeepRecords
#> [1] 4
#>
#> $nCyclesToRun
#> [1] 7
#>
#> $selCritPipeAdv
#> function (records, candidates, bsp, SP)
#> {
#> phenoDF <- framePhenoRec(records, bsp)
#> if (!any(candidates %in% phenoDF$id)) {
#> crit <- runif(length(candidates))
#> }
#> else {
#> crit <- iidPhenoEval(phenoDF)
#> crit <- crit[candidates]
#> }
#> names(crit) <- candidates
#> return(crit)
#> }
#> <bytecode: 0x7f971ece5108>
#> <environment: namespace:AlphaSimHlpR>
#>
#> $selCritPopImprov
#> function (records, candidates, bsp, SP)
#> {
#> phenoDF <- framePhenoRec(records, bsp)
#> if (!any(candidates %in% phenoDF$id)) {
#> crit <- runif(length(candidates))
#> }
#> else {
#> crit <- iidPhenoEval(phenoDF)
#> crit <- crit[candidates]
#> }
#> names(crit) <- candidates
#> return(crit)
#> }
#> <bytecode: 0x7f971ece5108>
#> <environment: namespace:AlphaSimHlpR>
#>
#> $analyzeInbreeding
#> [1] 0
#>
#> $chkReps
#> SDN CET PYT
#> 1 1 1
#>
#> $checks
#> NULL
#>
#> $plotCosts
#> SDN CET PYT
#> 1 8 14
#>
#> $perLocationCost
#> [1] 1000
#>
#> $crossingCost
#> [1] 0.2
#>
#> $qcGenoCost
#> [1] 1.5
#>
#> $wholeGenomeCost
#> [1] 10
#>
#> $develCosts
#> [1] 60
#>
#> $genotypingCosts
#> CET
#> 862.5
#>
#> $trialCosts
#> [,1]
#> [1,] 2645
#>
#> $locationCosts
#> [1] 2000
#>
#> $totalCosts
#> [,1]
#> [1,] 5567.5
nReplications <- 3
bsp$nCyclesToRun <- 6Run a simple breeding scheme for 6 cycles
Replicate a very simple breeding program 3 times.
replicRecords <- lapply(1:nReplications, runBreedingScheme, nCycles=bsp$nCyclesToRun, initializeFunc=initFuncADChk, productPipeline=prodPipeFncChk, populationImprovement=popImprov1Cyc, bsp)
#> ****** 1
#> [1] "initFuncADChk deprecated. Please use initializeScheme"
#> 1 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 2 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 3 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 4 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 5 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 6 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#>
#> ****** 2
#> [1] "initFuncADChk deprecated. Please use initializeScheme"
#> 1 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 2 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 3 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 4 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 5 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 6 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#>
#> ****** 3
#> [1] "initFuncADChk deprecated. Please use initializeScheme"
#> 1 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 2 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 3 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 4 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 5 [1] "prodPipeFncChk deprecated. Please use productPipeline"
#> 6 [1] "prodPipeFncChk deprecated. Please use productPipeline"Calculate the means of the breeding programs
And plot them out
plotData <- plotRecords(replicRecords)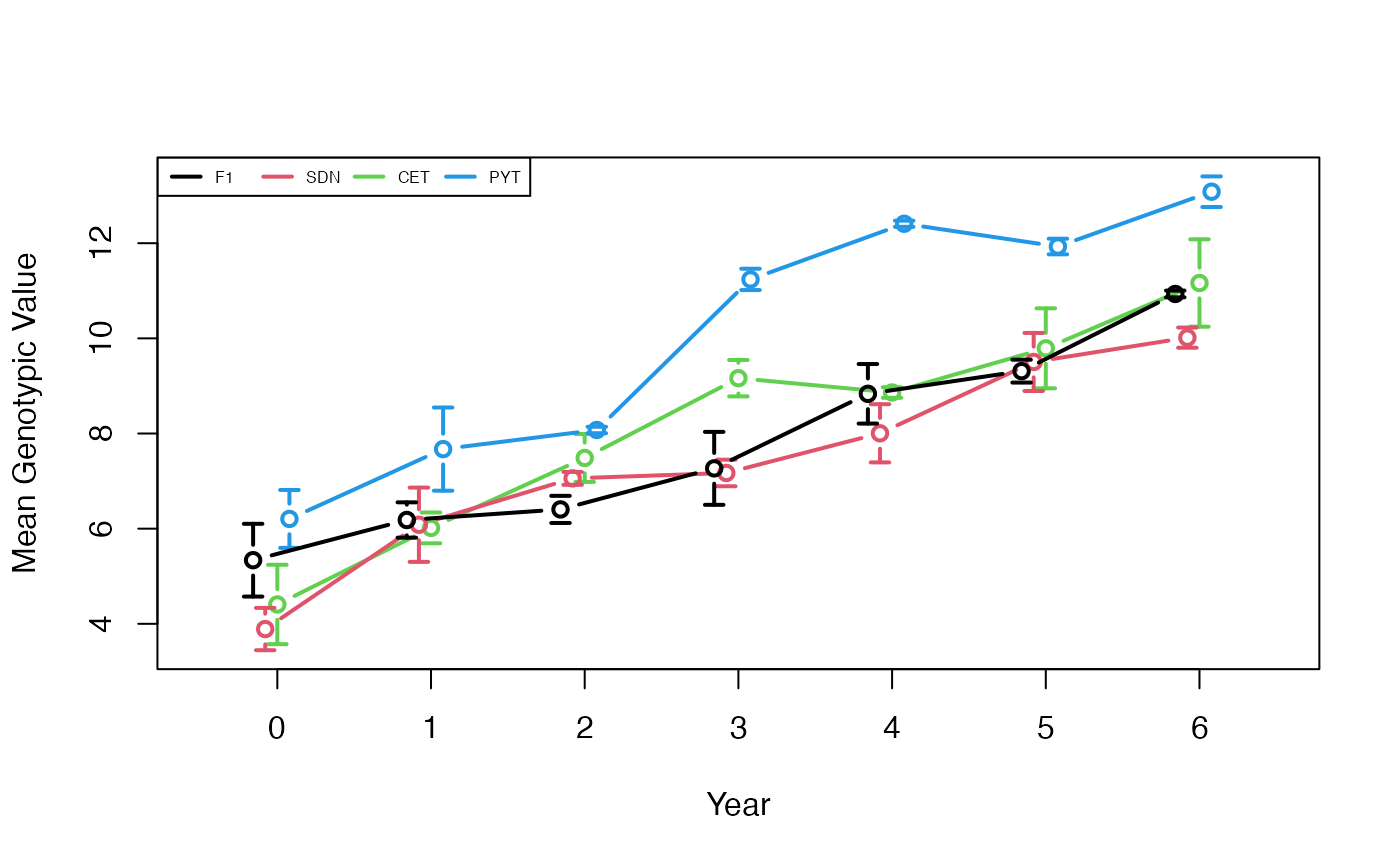
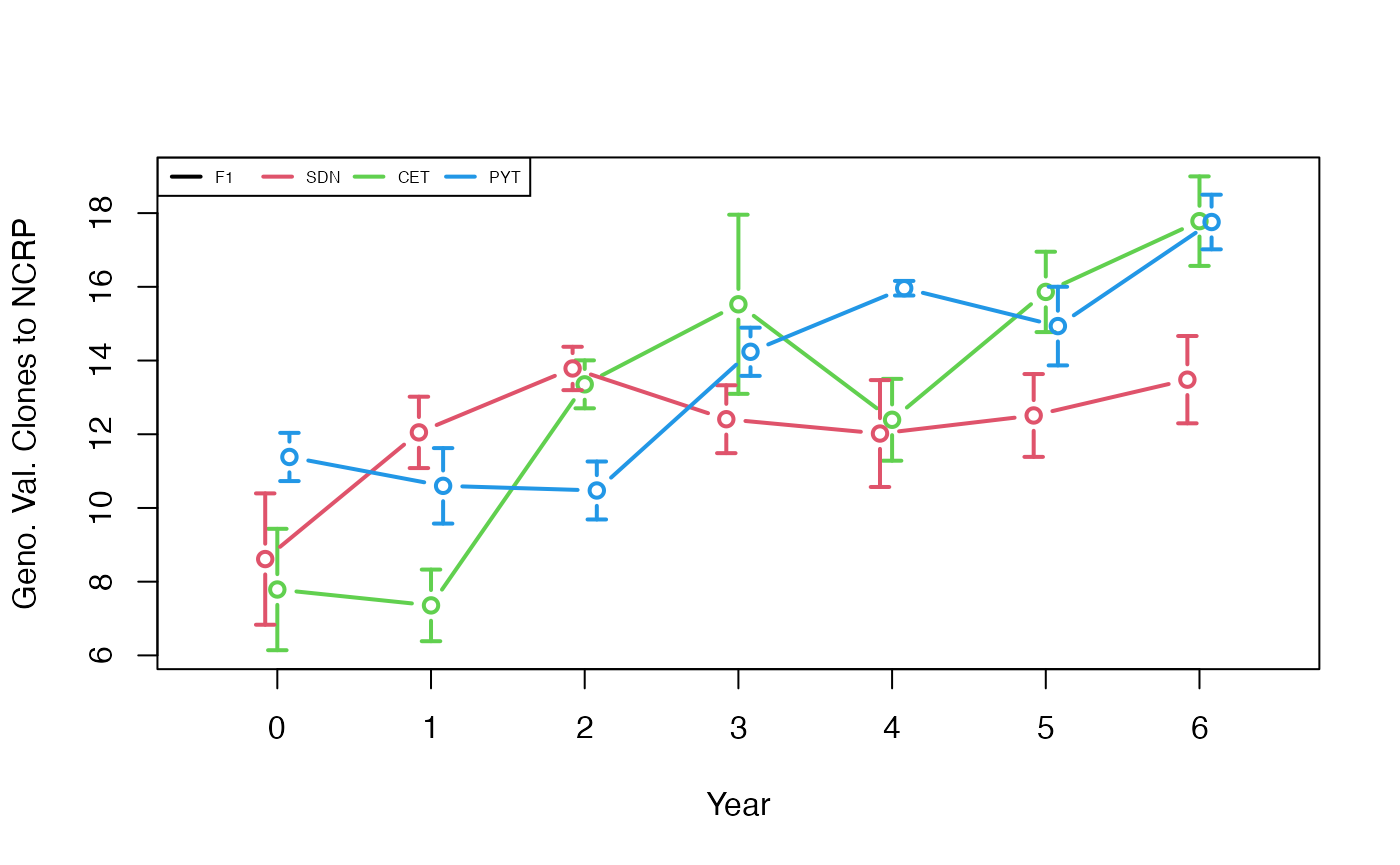
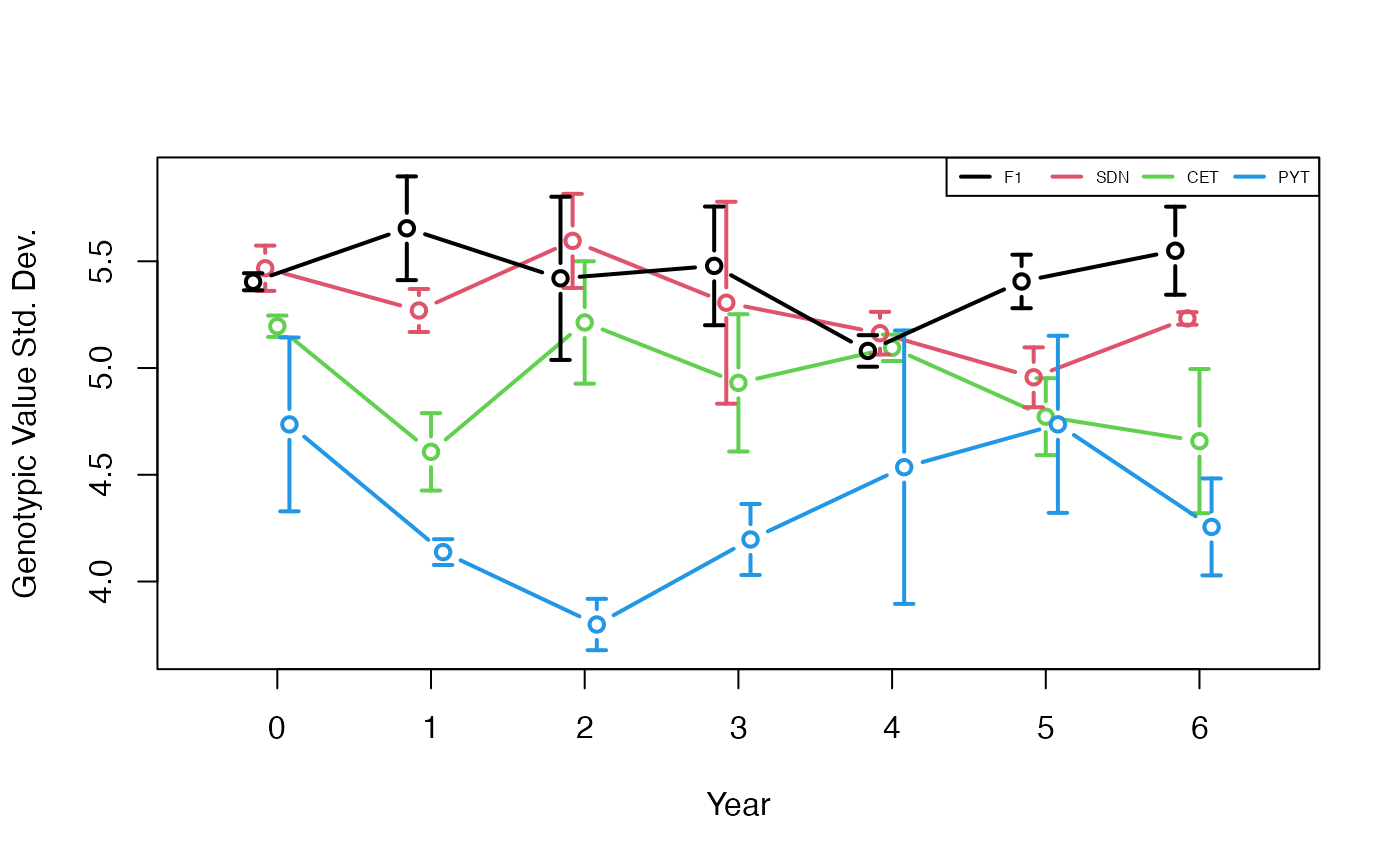
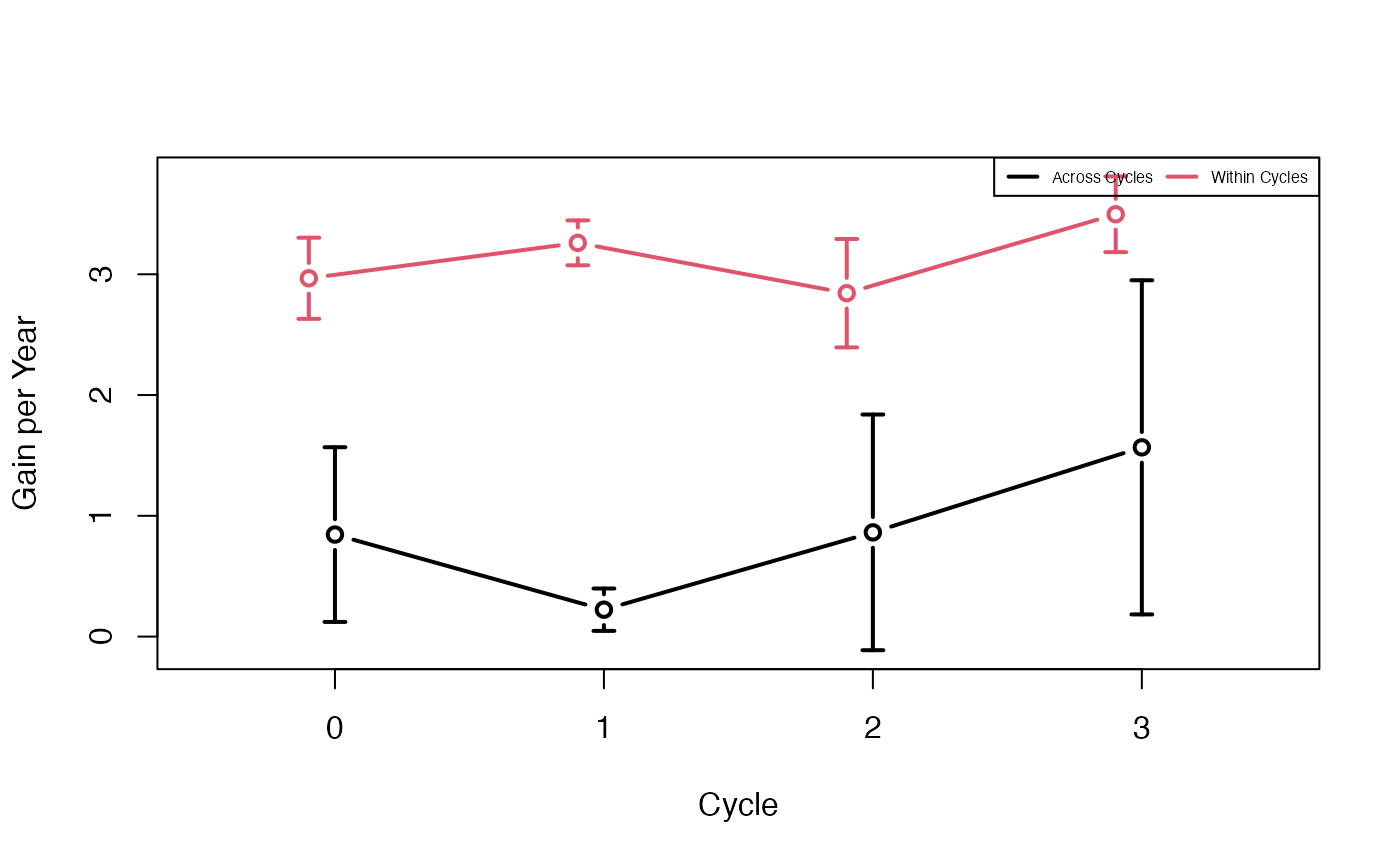
meanMeans <- tapply(plotData$genValMean, list(plotData$year, plotData$stage), mean)
meanMeans <- meanMeans[,c("F1", bsp$stageNames)]
stdErrMeans <- tapply(plotData$genValMean, list(plotData$year, plotData$stage), std.error)
stdErrMeans <- stdErrMeans[,c("F1", bsp$stageNames)]
print(meanMeans)
#> F1 SDN CET PYT
#> 0 5.337219 3.889841 4.406701 6.204772
#> 1 6.181973 6.082486 6.015928 7.672369
#> 2 6.404645 7.056919 7.486252 8.074156
#> 3 7.267598 7.170380 9.163059 11.239164
#> 4 8.834539 8.004504 8.862132 12.409074
#> 5 9.311167 9.503764 9.790297 11.930509
#> 6 10.933441 10.015411 11.163668 13.082785
print(stdErrMeans)
#> F1 SDN CET PYT
#> 0 0.76461925 0.4447262 0.8345590 0.60881823
#> 1 0.37134206 0.7806114 0.3237561 0.87436656
#> 2 0.28523807 0.1354334 0.5034539 0.06765403
#> 3 0.76656060 0.2791706 0.3825326 0.22424016
#> 4 0.62593831 0.6114918 0.1112071 0.06511340
#> 5 0.24034729 0.6103133 0.8408421 0.16446667
#> 6 0.07086069 0.2125570 0.9178999 0.32168814